Similar Conditions
Getting a diagnosis for any rare disease can be frustrating. Among the K-T population, rediagnosis has been a common occurrence. K-T is the oldest and most well known of the combined vascular anomalies and for many years was an umbrella diagnosis. With more recent advances like those listed below, many once diagnosed with K-T are finding new, more precise descriptors for their condition.
- gradual long term shift toward more focus on specialties in medicine
- vast improvement and advancement in medical imaging technology
- ISSVA classification redefined the way clinicians view these conditions.
- CLOVES, a condition identified in 2007, more aptly defined some cases previously diagnosed as K-T
- RASA1 mutation was identified in many Parkes Weber cases, allowing it to be more easily differentiated from K-T
- AKT1 gene was identified in Proteus patients
- K-T was identified as a PIK3CA related overgrowth syndrome (PROS) in April 2015. Among the PROS identified are CLOVES, FAVA (fibroadipose vascular anomaly), M-CM, lymphatic malformations. Overlap in these conditions often occur.
Diagnosis of combined malformations is difficult. Similar conditions can have symptoms that might not be apparent at an early age but become more prominent with puberty - the onset of the majority growth period plus accompanying hormonal changes.
Washington University Genomics and Pathology Services offers genetic (SOMA) testing that can identify a PIK3CA mutation. Identification of a specific PIK3CA condition beyond gene identification is not possible at this time.
Conditions that might have some similarity to K-T syndrome are:
Parkes-Weber Syndrome (PWS, PKWS)
Identified gene mutation: RASA1
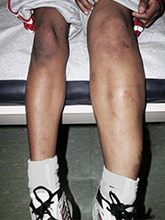
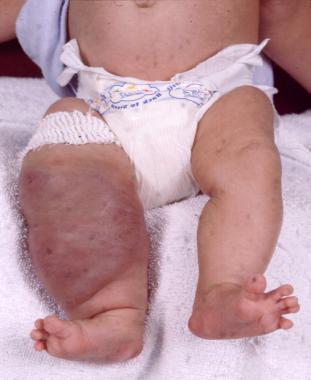
Parkes Weber syndrome is a disorder of the vascular system characterized by vascular abnormalities known as capillary malformations and arteriovenous fistulas (AVFs), which are present from birth. The capillary malformations increase blood flow near the surface of the skin. They usually look like large, flat, pink stains on the skin, and because of their color are sometimes called "port-wine stains." In people with Parkes Weber syndrome, capillary malformations occur together with multiple micro-AVFs, which are tiny abnormal connections between arteries and veins that affect blood circulation. These AVFs can be associated with life-threatening complications including abnormal bleeding and heart failure.
Another characteristic feature of Parkes Weber syndrome is overgrowth of one limb, most commonly a leg. Abnormal growth occurs in bones and soft tissues, making one of the limbs longer and larger around than the corresponding one.
Some vascular abnormalities seen in Parkes Weber syndrome are similar to those that occur in a condition called capillary malformation-arteriovenous malformation syndrome (CM-AVM). CM-AVM and some cases of Parkes Weber syndrome have the same genetic cause. (1)
Parkes Weber and capillary malformation-arteriovenous malformations are very often incorrectly identified as K-T. The conditions are very similar in appearance but unlike K-T, Parkes Weber includes arteriovenous malformations that affect the health of the heart. (1)
Management of Parkes Weber is similar to K-T in the areas of skin care, exercise, diet, most orthopedic issues, and lymphedema management. Additional complications from Parkes Weber include the risk of high-output cardiac failure. Bleeding is a common complication with both K-T and Parkes Weber, and parents or caregivers should be familiar with first aid instructions.
image
{kind=link}
1) Boston Children's Conditions and Treatments
2) NIH Genetic Home Reference, Parkes Weber syndrome
Capillary malformation-arteriovenous malformation (CM-AVM)
Identified gene mutation: RASA1
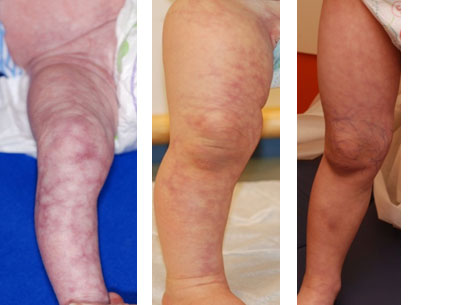
CM-AVM is characterized by capillary malformations (CMs), which are composed of enlarged capillaries that increase blood flow near the surface of the skin. These malformations look like multiple small, round, pink or red spots on the skin. In most affected individuals, capillary malformations occur on the face, arms, and legs. These spots may be visible from birth or may develop during childhood. By themselves, capillary malformations usually do not cause any health problems.
In some people with CM-AVM, capillary malformations are the only sign of the disorder. However, other affected individuals also have more serious arteriovenous malformations (AVMs) and arteriovenous fistulas (AVFs). AVMs and AVFs are abnormal connections between arteries, veins, and capillaries that affect blood circulation. Depending on where they occur in the body, these abnormalities can be associated with complications including abnormal bleeding, migraine headaches, seizures, and heart failure. In some cases the complications can be life-threatening. In people with CM-AVM, complications of AVMs and AVFs tend to appear in infancy or early childhood; however, some of these vascular abnormalities never cause any symptoms. (1)
1) NIH Genetic Home Reference, CM-AVM
Congenital Lipomatous Overgrowth, Vascular Malformations, Epidermal Nevis, Spinal/Skeletal Anomalies/Scoliosis syndrome (CLOVES)
CLOVES Syndrome is a recently described overgrowth syndrome with complex vascular anomalies. CLOVES stands for Congenital, Lipomatous Overgrowth, Vascular malformations, Epidermal nevi and Scoliosis/Skeletal/Spinal anomalies. The syndrome was described independently by Saap et al. and Alomari [1,2]. and shares a random mosaic mutation in the PIK3CA gene. CLOVES includes
- lipomatous truncal masses
- overgrowth or deformities in their arms/hands and/or their legs/feet
- vascular and lymphatic anomalies; spinal AVM
- skin abnormality
- scoliosis or other spinal problem
Not all signs are present in most patients and the condition can vary greatly in severity and percentage of body involvement.
1) Sapp JC, Turner JT, van de Kamp JM, van Dijk FS, Lowry RB, Biesecker LG. 2007. Newly delineated syndrome of congenital lipomatous overgrowth, vascular malformations, and epidermal nevi (CLOVE syndrome) in seven patients. AmJ Med Genet Part A 143A: 2944-2958.
2) Alomari AI. 2009. Characterization of a distinct syndrome that associates complex truncal overgrowth, vascular, and acral anomalies: A descriptive study of 18 cases of CLOVES syndrome. Clin Dysmorphol;18:1-7.
image
CLOVES Syndrome Community
{kind=link}
{kind=link}
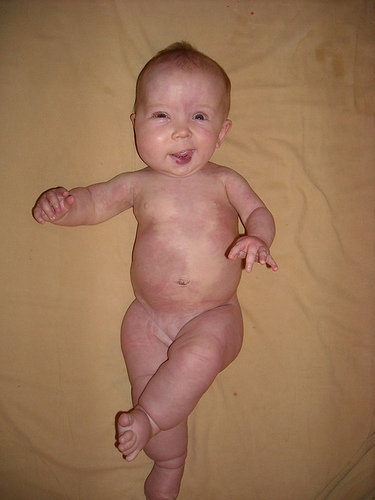
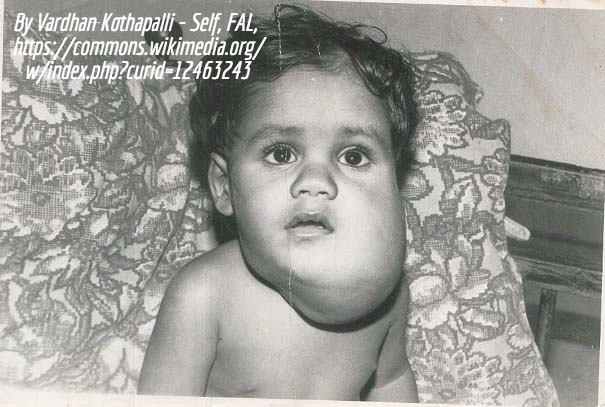
Macrocephaly capillary malformation syndrome (M-CM, MCAP)
Macrocephaly-capillary malformation is a rare, multiple malformation syndrome characterized by a spectrum of anomalies including primary megalencephaly, prenatal overgrowth, brain and body asymmetry, cutaneous vascular malformations, digital anomalies consisting of syndactyly with or without postaxial polydactyly, connective tissue dysplasia involving the skin, subcutaneous tissue, and joints, and cortical brain malformations, most distinctively polymicrogyria.
Generally, individuals with KTS do not have macrocephaly and do not have significant developmental delay or neurologic problems but in some cases M-CM and K-T have enough overlap that a diagnosis is not simple.
image
M-CM Network
{kind=link}
{kind=link}
Proteus Syndrome
Proteus syndrome is characterized by progressive, mosaic, segmental overgrowth that can affect any organ or tissue in the body
image
Proteus Syndrome Foundation
{kind=link}
1) Biesecker, L. G. The Multifaceted Challenges of Proteus Syndrome. JAMA 285, 2240–2243 (2001).
2) Malamitsi-Puchner, A., Dimitriadis, D., Bartsocas, C. & Wiedemann, H. R. Proteus Syndrome: Course of a Severe Case. Am. J. Med. Genet. 35, 283–285 (1990).
3) Iqbal J. et al. Case Report:Morphological Characterization of the Breast in Proteus Syndrome Complicated by Ductal Carcinoma In Situ. Ann. Clin. Lab. Sci. 36, 469–474 (2006).
4) Turner, J. T., Cohen, M. M., Jr. & Biesecker, L. G. Reassessment of the Proteus syndrome literature: Application of diagnostic criteria to published cases. Am. J. Med. Genet. 130A, 111–1
5) Slavotinek, A. M., Vacha, S. J., Peters, K. F. & Biesecker, L. G. Sudden death caused by pulmonary thromboembolism in Proteus syndrome. Clin. Genet. 58, 386–389 (2000).
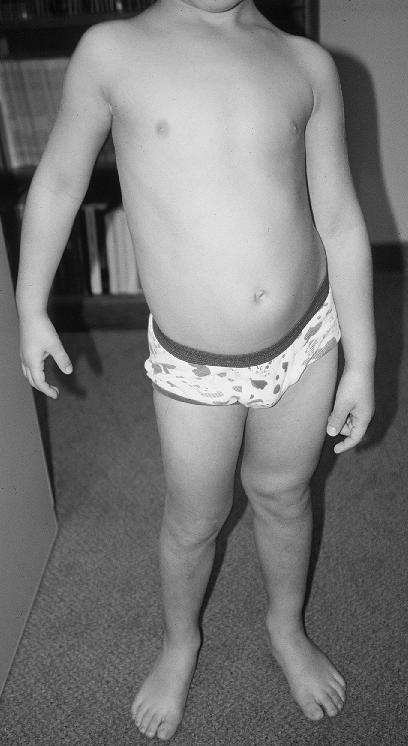
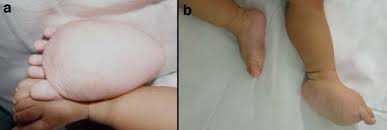
Diffuse Capillary Malformation with Overgrowth (DCMO)
DCMO, recently described by Margaret S. Lee et al,[1] has been proposed as an independent entity within the wide spectrum of vascular abnormalities associated with overgrowth. It is distinguished by erythematous-purplish stains, with a narrow network morphology than can go with a more homogeneous or plain macules. It has a diffuse distribution, extending minimum beyond one anatomic region, and neither atrophy nor ulcers are present. It has been observed a midline sharply demarcation on the abdomen, but never on the back. The associated overgrowth is a diffuse proportional enlargement of a body region, most commonly of a limb, that does not progress, accordingly vascular or infectious complications will not take place. The skin vascular malformations frequently lighten over the first several months, however less intense than CMTC.
This recently proposed designation describes patients with an extensive, diffuse, reticulate capillary malformation and variable, but proportionate, hypertrophy without any major complications. A reticulate capillary malformation is defined as networklike, blotchy, nonuniform in color, and without distinct borders. This is in contradiction to the darker "geographic" stains observed in KTS. These patients do not fit the diagnostic criteria of the other disorders marked by capillary malformations and overgrowth such as KTS or Parkes-Weber syndrome. Patients still require periodic evaluation to monitor for leg length discrepancy. They exhibit normal neurologic development and proportionate overgrowth rather than progressive, disproportionate symmetry.
image
1)f Lee MS, Liang MG, Mulliken JB. Diffuse capillary malformation with overgrowth: a clinical subtype of vascular anomalies with hypertrophy. J Am Acad Dermatol. 2013;69:589–594.
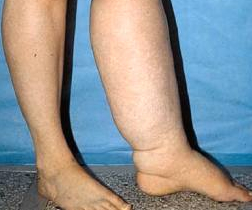
Lymphedema
Lymphedema, also known as lymphatic obstruction, is a condition of localized fluid retention and tissue swelling caused by a compromised lymphatic system, which normally returns interstitial fluid to the thoracic duct and then the bloodstream. The condition can be inherited or can be caused by a birth defect, though it is frequently caused by cancer treatments, and by parasitic infections. Though incurable and progressive, a number of treatments can ameliorate symptoms. Tissues with lymphedema are at high risk of infection.
image
National Lymphedema Network
{kind=link}
Lymphatic malformation
Lymphatic malformations are a type of vascular malformation. They are rare non-malignant masses consisting of fluid-filled channels or spaces thought to be caused by the abnormal development of the lymphatic system. The lymphatic system normally collects excess fluid from the tissues and transports it through a series of small vessels back into the venous system. A lymphatic malformation will slow the transfer of fluid through these vessels. The excess fluid accumulates and dilates the vessels, resulting in a swelling of the affected area and sometimes more extensive enlargement of soft tissue and bones. These malformations are usually apparent at birth or by two years of age.
The lymphatic system normally collects excess fluid from the tissues and transports it through a series of small vessels back into the venous system. With a lymphatic malformation, however, transfer of this fluid through these vessels is slowed. The excess fluid accumulates and dilates the vessels, resulting in a swelling of the affected area and sometimes in more extensive enlargement of soft tissues and bones.(1)
These malformations are usually apparent at birth, or by two years of age. Lesions may be superficial or deep, and localized or diffuse. They steadily increase in size, although some enlarge more rapidly than others. Conditions such as infection or trauma can result in sudden but temporary enlargement.
Lymphedema is a form of lymphatic malformation.(2)
image1
image 2
Kennedy's Cause
{kind=link}
{kind=link}
1) Cincinnati Children's Hospital
2) Boston Children's Hospital
Venous malformation
Venous malformations are abnormally formed and dilated veins that can be either superficial or deep. They are the most common kind of vascular lesion. They are usually present at birth but some might not be apparent until later.
Venous malformations enlarge slowly over time but can experience rapid growth when some trauma occurs, such as surgery, infection, or some hormonal interference (puberty, pregnancy, menopause, or imbalance). They can be present in the skin, mucous membrane or in any organ system.
image
NOVA
Vascular Birthmarks Foundation (VBF)
{kind=link}
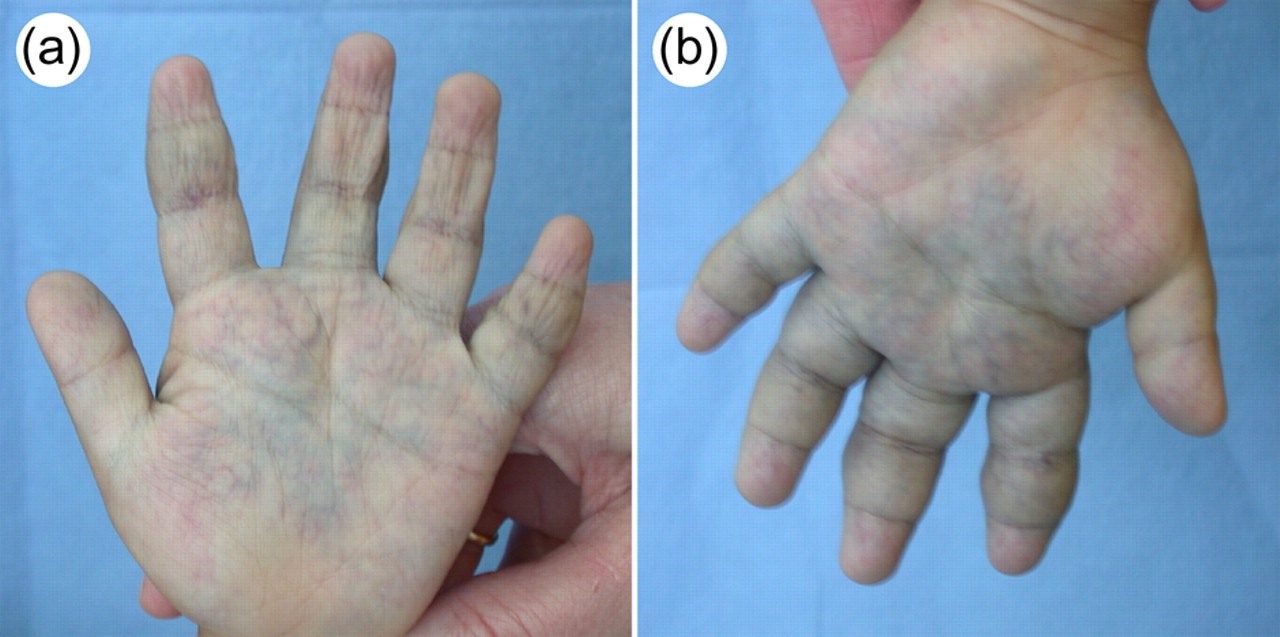
Hemihyperplasia/Hemihypertrophy
Hemihyperplasia, formerly called hemihypertrophy, is a rare congenital disorder in which one side of the body grows more than other, causing asymmetry. In a normal cell, there is a mechanism that turns off growth once the cell reaches a certain size. However, in hemihypertrophy, the cells on one side aren’t able to stop growing. This causes the body to continue growing or enlarge abnormally.
image
{kind=link}
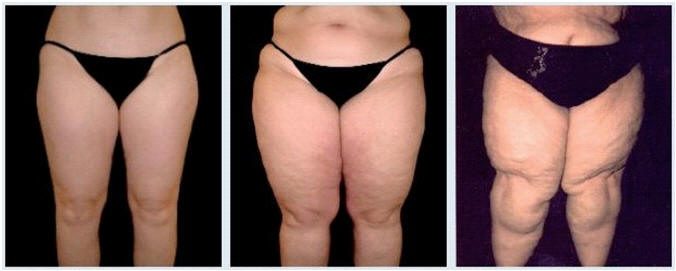
Lipedema
Lipedema, an abnormality of adipose tissue, is often confused for lymphedema or obesity. Few in the medical community are even familiar with this condition that is now estimated to affect up to 11% of adult women. Its onset often accompanies periods of hormonal change such as puberty, pregnancy or menopause. Typically a person suffering from lipedema is much heavier on the lower half of their body and if they do lose weight it is from their upper body. The legs may be painful to touch and bruise easily.(1)
image
{kind=link}
Infantile hemangioma
Infantile hemangiomas are benign vascular tumors of childhood, characterized by endothelial cell proliferation. They are the most common soft-tissue tumors of childhood, occurring in 3% to 10% of the population. The lesions are usually not detectable at birth but appear during the first 4 to 6 weeks of life. All infantile hemangiomas exhibit characteristic evolution with early rapid growth (proliferation) followed by a stabilization period and a slow spontaneous involution. The growth phase can extend up to 6 months of age, followed by an involution phase over a period of 3 to 7 years.
image
National Organization for Vascular Anomalies (NOVA)
Vascular Birthmarks Foundation (VBF)
{kind=link}
Kaposiform Hemangioendothelioma (KHE)
Kaposiform hemangioendothelioma (KHE) is an infrequent and aggressive childhood tumor, involving the blood vessels. These tumors may be located on the skin surface, or deep inside the body tissues
KHE manifests before, or shortly after birth and is often linked with Kasabach-Merritt phenomenon. This phenomenon is a condition where vascular tumors destroy blood platelets
KHE is commonly found in the retroperitoneum space (near abdominal cavity) and beneath the skin surface, head, neck and torso.
image
{kind=link}
Lipofibromatosis
A rare, slow-growing painless tumor of infancy and childhood that usually affects the distal extremities. It is characterized by the presence of alternating bands of mature adipose tissue and spindle-cell fibrous tissue, without destruction of the adipose tissue architecture. It does not metastasize but the rate of local recurrence is high.
Average age of diagnosis is 1 year, most cases range from 11 days to 12 years of age. There is a 2:1 predominance of male to female. It is commonly found on the hands and feet, with some cases on the trunk, neck and head. The tumor measures 1-3 cm average, and is rarely over 5 cm. Recurrence rate after treatment is most common.
image
{kind=link}
Cutis Marmorata Telangiectatica Congenita (CMTC)
CMTC is characterized by discolored patches of the skin due to prominent blood vessels.The result is like cutis marmorata (pinkish blue mottled or marbled appearance) but is much more pronounced. Other vascular malformations such as hemangiomas, salmon patches and varicose veins can also occur in patients with CMTC. The cause is unknown and while it is often an isolated finding, there are risks of other associated anomalies including glaucoma and limb asymmetry (most often hemihypoplasia, or undergrowth of the affected limb).
image
{kind=link}
Capillary malformations, congenital (CMC)
A capillary malformation (commonly referred to as a port-wine stain), is a flat, sharply defined vascular stain of the skin. It may cover a large surface area or it may be scattered and appear as little islands of color. It can be anywhere on the body and in more than one place on the body, but is most commonly seen in the head/neck region. (1)
Although the number of blood vessels in a capillary malformation is normal, the diameter of the affected vessels is much larger than that in normal vessels; this enlargement results in increased blood flow. Because the vessels are close to the surface, this increased flow gives the skin its pink to purple appearance.
As the child grows, the affected blood vessels will continue to enlarge and thicken, causing the color of the lesion to darken. Over time, the clusters of tiny, dilated venules (small vessels that collect blood from the capillary junctions and join to form veins) give a lumpy appearance to the skin. The period of time over which this progression occurs varies greatly from person to person, and may even be delayed until ages 40, 50, or 60.
Capillary malformations on the forehead and upper eyelid can be associated with lesions of the brain and eye (see Sturge Weber).
Capillary malformations in the skin over the spine can be associated with Cobb syndrome, which involves the spine and/or meninges (tissue covering the spinal cord). Capillary malformations in this area can also be associated with other abnormalities of the spine, and should be investigated with an ultrasound or MRI.
Capillary malformations located in the middle of the face (forehead, nose, upper lip) can be associated with vascular abnormalities of the brain, and should also be investigated.
A group of capillary vascular lesions seen primarily in newborn is often confused with capillary malformations. When these birthmarks appear on the forehead, eyelids, nose or upper lip, they are commonly called "angel kisses." When located on the back of the neck, they are commonly called "stork bites." These lesions usually fade by 1 year of age and do not require treatment.
Birthmark Support Group, UK
Vascular Birthmark Foundation
NOVA
(1) Description reprinted from Cincinnati Children's Hospital
Sturge-Weber
Sturge-Weber syndrome is characterized by an intracranial vascular anomaly, leptomeningeal angiomatosis, most often involving the occipital and posterior parietal lobes. The most common symptoms and signs are facial cutaneous vascular malformations (port-wine stains), seizures, and glaucoma. Stasis results in ischemia underlying the leptomeningeal angiomatosis, leading to calcification and laminar cortical necrosis. The clinical course is highly variable and some children experience intractable seizures, mental retardation, and recurrent stroke-like episodes. Sturge Weber syndrome is caused by a somatic activating mutation in GNAQ.
image
Sturge Weber Foundation
{kind=link}
PTEN-Related Hamartoma Syndromes
PTEN is a gene that, when altered, can cause different disorders that are characterized by the development of noncancerous tumor-like growths called hamartomas. Together, the disorders caused by PTEN are called PTEN-related hamartoma syndromes and are summarized below. Some but not all cases of the following conditions are due to identifiable changes in the PTEN gene.These conditions are included because some may bear a superficial appearance similar to K-T.
- Bannayan-Riley-Ruvalcaba syndrome (BRRS) is characterized by macrocephaly, hamartomas in the intestines (intestinal hamartomatous polyposis), lipomas and pigmented speckling on the skin of the penis in males. (images) (more information)
- Cowden syndrome is a rare disorder that causes multiple hamartomas and an increased risk of certain cancers of the thyroid, endometrium (lining of the uterus) and breast. Hamartomatous growths can also occur on the skin, intestinal tract and mucous membranes (such as the lining of the mouth and nose). Affected individuals often have macrocephaly. (image)
- Proteus-Like syndrome (PLS). Individuals with PLS have been found to have mutations in PTEN and clinically resemble Proteus syndrome. This is a variable group of clinical entities and diagnosis of an individual can be challenging unless a provider is experienced with the condition.
{kind=link}
{kind=link}
There are many different kinds of conditions that are classified as vascular anomalies. If you would like to learn more about different types of vascular malformations, read ISSVA's updated classification information at the ISSVA website, or more detailed information in the July 2015 Pediatrics journal article Vascular Anomalies Classification: Recommendations from the International Society for the Study of Vascular Anomalies.
Page last updated December 21, 2017